Aligning Selected Elements
All elements which will be selected in the Alignment List
will be aligned to a reference element. This reference element can
be set by the user or will be chosen automatically as described
above. To perform an alignment, select two or more elements in the
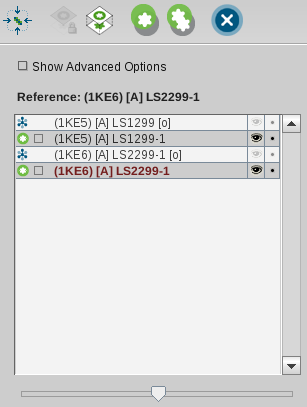
Alignment List. This should look like the following image where
pharmacophores of ligands of complexes 1ke5 and 1ke6 were selected
and 1ke6 was set to be the reference element.
After clicking the icon
Align Selected Elements
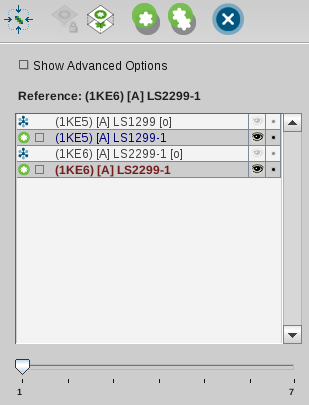
LigandScout will calculate one or several alignments and present
them to the user as illustrated in the image below. The activated slider
indicates that there are several alignments available. Information about
each alignment are found in the
Alignment Details View
, e.g.,
the associated RMS (root mean squared) value of the
valid matched feature deviances is shown as well as the number of
valid matched features.
There are cases where LigandScout will provide just a
single alignment:
-
Three or more elements will be aligned to each
other
-
A perfect alignment was
found (usually a molecule to its own pharmacophore,
or aligning two instances of the same structure)