|
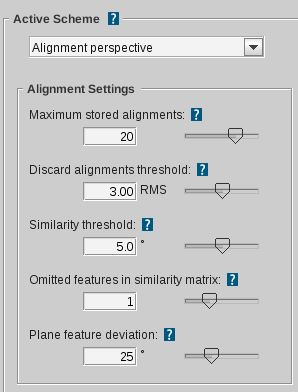
Active scheme
|
Specifies the settings scheme for alignments in
the screening process (Match all query, Fragment screening), ligand-based model creation or Alignment Perspective.
|
|
Maximum stored alignments
|
Defines the maximum number of stored alignments. In case there will
be more alignments during the execution of the alignment algorithm, only the best
will be kept and the others will be discarded. Note: The so called
Pharmacophore-Fit scoring function (considers number of features and RMS) is used.
The scoring function can be customized only in the
ligand-based settings
to generate
ligand-based pharmacophores. (default for Alignment Perspective and ligand-based model creation: 20; Screening: 1 )
|
|
Discard alignments threshold
|
Defines the maximum allowed RMS (root mean squared)
deviation of structures to be considered alignable. Alignments
exceeding this limit are rejected. The RMS calculation is based
on valid matched feature pairs of both structures.
(default: 3.00 RMS)
|
|
Similarity threshold
|
For every generated alignment a rotation is stored.
Depending on the deviation of the three unit vectors under this
mapping an alignment will be discarded if it is too similar to a
previous one. Certainly, it will be checked which alignment has
the better RMS value and this one will be kept.
(default: 5.0)
|
|
Omitted features in similarity matrix
|
Defines how many features can be omitted during
alignment. Alignments will be generated by combinatorially
excluding the specified number of features. This means that the
more features are left out, the longer the generation process
will take (NOTE: exponential growth). (default: 1)
|
|
Plane feature deviation
|
Defines the maximum deviation of the normal vectors
of two plane features to be included in an alignment. If the
angle between the normal vectors exceeds the specified value,
there will be no valid match between these two plane features.
(default: 25°)
|