
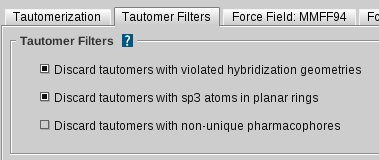
Tautomerization Settings
allow for
the customization of various parameters influencing ligand tautomer
enumeration and filtering.
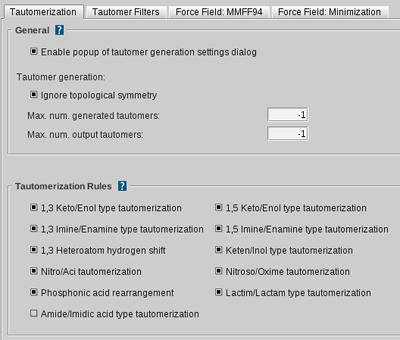
| General | Effect |
|---|---|
| Enable popup of tautomer ceneration settings dialog | Enables the settings dialog of the tautomer generation. (default: enabled) |
| Ignore topological symmetry | If not checked, topologically identical ligand tautomers are discarded even if they are different in 3D structure. (default: enabled) |
| Max. num. generated tautomers | Specifies the maximum number of unfiltered tautomers to generate. A value of 0 or -1 means no limit. (default: -1) |
| Max. num. output tautomers | Specifies the maximum number of filtered output tautomers to generate. A value of 0 or -1 means no limit. (default: -1) |
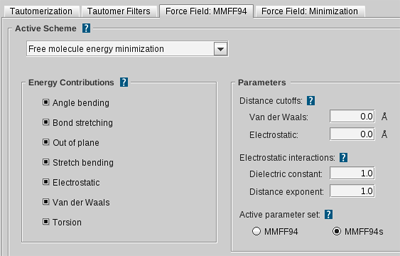
MMFF94 and Minimization Settings
allow for the customization of various parameters influencing MMFF94
forcefield energy calculations and the conjugate gradients algorithm
used for structure optimization.
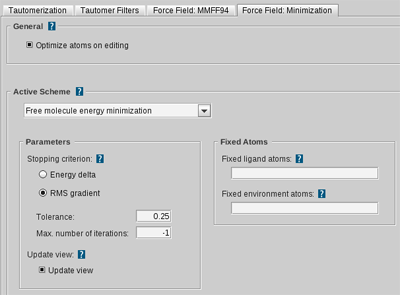
| General | Effect |
|---|---|
| Optimize atoms on editing | If selected, the edited atoms and bonds (type change, deletion, addition) are optimized automatically. (default: enabled) |