
LigandScout's menu bar consists of:
Displays the
Open File
dialog (CTRL+O). Various file formats presenting chemical
structure information (e.g.: *.pdb, *.sdf, ...) can be imported to
LigandScout. If you open new data, all current data is closed before loading the new one.
This command allows the
injection
of additional molecules
into your shown data (binding site
and/or pharmacophore) in the
Structure-Based Modeling Perspective
(CTRL+I).
Save to Repository
(CTRL+S) stores active sites and macromolecule information in your Local Repository.
Save as File
(CTRL+SHIFT+S)
enables you to
save/export
your macromolecule and ligand modifications as well as your
pharmacophore models. Depending on the file format, check boxes in
the
Save File dialog
let you choose advanced export settings and the items (ligand,
environment, pharmacophore) you want to save/export.
The export settings are sensitive according to the current data
(e.g.: if no pharmacophore is present then related settings are disabled).
Closes the shown data in the current view (CTRL+W). In the
Structure-Based Modeling Perspective
LigandScout gives feedback and asks for confirmation, in case there
is modified or unsaved data.
Loads the previously exported SES file which contains
data of the Local Repository, Alignment Perspective, Ligand-Based Modeling Perspective
and Bookmark View.
Stores the current data of the Local Repository, Alignment Perspective, Ligand-Based Modeling Perspective
and Bookmark View in a single session file (SES file).
Stores the current data of the Local Repository, Alignment Perspective, Ligand-Based Modeling Perspective
and Bookmark View in a single session file. Selecting your named session reloads
all the data that was previously stored.
Deletes one of the available named session files from disk (
irrevocable operation).
Deletes the current PDB complex from your Local Repository
(CTRL+ALT+SHIFT+D). LigandScout
will reload the original data from the PDB server if requested.
Import Repository
(CTRL+ALT+SHIFT+I) allows you to import a zipped PMZ database
(*.zip) either locally or to your default LigandScout data search
path. The use of a PDB data repository drastically accelerates the
ligand-macromolecule complex initialization, since LigandScout uses
the structural information stored in the repository instead of
downloading the file from the PDB server and interpreting it.
Export Repository
(CTRL+ALT+SHIFT+E) allows you to export a zipped PMZ database
(*.zip) either from the Local Repository or from your Shared Repository.
Clear Local Repository
(CTRL+ALT+SHIFT+C) erases the local PDB data storage.
Imports a previously exported set of alignments
(CTRL+ALT+V). Using this functionality will directly
add all stored molecules and pharmacophores to the
Alignment Perspective
.
In case the
Alignment Perspective
already contains elements, a dialog will pop up and ask if the old
elements should be deleted before adding the new ones.
Export
(CTRL+ALT+B) will export all molecules and
pharmacophores which are currently listed in the
Alignment Perspective
.
Print Preview
(CTRL+ALT+P) shows the report
that is printed with the
Print
command. Depending
on the current view,
Print Preview
will show either a 3D image of the macromolecule if the application
is in the Macromolecule View or it will show a report that will be
printed with the
Print
command if the application
is in the
Active Site View
.
In the Macromolecule View the result will look like:
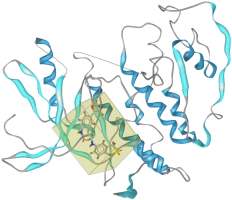
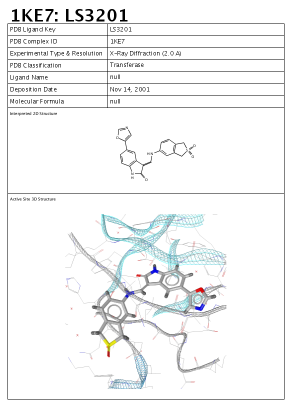
In the Active Site View the report which will show up
contains a 3D image of the active site, a 2D depiction of the
ligand as well as PDB complex meta data consisting of:
-
PDB ligand key
-
PDB complex ID
-
Experimental type and resolution
-
PDB classification
-
Ligand name
-
Deposition date
-
Molecular formula
Here is an example of this report:
Print
(CTRL+P)
enables you to print out the current complex as a report containing
a 3D image of the active site, a 2D depiction of the ligand as well
as PDB complex meta data consisting of: PDB ligand name,
complex identifier, PDB classification, etc.
LigandScout uses white background for printing.
If you want to export the 2D and 3D View of your shown elements
as images, for instance molecule alignments or single pharmacophore,
select the
Save
command in the
File
menu. LigandScout supports a
various list of image file types such as PNG, POV etc.
For more information please see
the section called “Exporting Files”
Restore Factory Defaults
(CTRL+SHIFT+F)
will delete your Local Repository, alignment and bookmark lists,
dictionaries and preference settings. In fact it will delete
everything which is contained in the folder
.inteligand-data
(the application's working directory).
LigandScout journalizes the eight most recent files
for fast and simple reaccess (CTRL+SHIFT+1...9).
Redo
(CTRL+Y)
is the counterpart of the
Undo
command and executes your previously reversed action.
Select: All
(CTRL+A)
picks all items (ligand, environment and pharmacophore) at once.
The items can be individually selected
by choosing “
Ligand
”, “
Environment
”
or “
Pharmacophore
”.
Invert Selection
inverts the current selection, i. e. this command selects all
elements without the currently selected elements.
Delete Selected Elements
(DEL)
will delete all elements contained in the current selection.
The
Preferences
(CTRL+SHIFT+P) allow you to customize LigandScout settings. This item can be
accessed via the
LigandScout
menu on Mac OSX platforms.
Calculates the score between the shown pharmacophore and the
ligands of the library. The pharmacophore score gives information
about the fit between a pharmacophore and a molecule. This score
takes into account the number of matched features times 10 and the
RMSD (Root Mean Square Deviation).
Calculate Score to Shown Pharmacophore (Without
Exclusion Volumes)
calculates the score between the
shown pharmacophore and the ligands of the library. The exclusion
volumes are not considered in score calculation.
Align and Calculate Score to Shown
Pharmacophore
first aligns the molecules of the library
with the shown
pharmacophore. Then the score is calculated. The
exclusion volumes
are included in this score calculation. If a
ligand extends into any
of the exclusion volumes the score is set
to -1.
Align and Calculate Score to Shown Pharmacophore
(Without Exclusion Volumes)
aligns first the molecules of the library with the shown
pharmacophore. Then the score is calculated. The exclusion volumes
are not included in the scoring process.
Calculate Standard Properties
calculates the standard properties of a molecule library. This
includes the calculation of Rel. TPSA, TPSA [P.Ertl], number of
rotatable bonds, acceptors, rings, cLogP, MolWt, heavy atoms and
donors.
Calculate Gaussian Shape Similarity Score
uses Gaussian functions for
analytically overlap calculations.
Calculate MMFF94 Energy
calculates the MMFF94 energy of molecules using the
settings scheme
"Library Molecule Energy Calculation" in the preferences.
Calculate MMFF94 Binding Enthalpy
calculates the MMFF94 binding enthalpy of molecules relative to the surrounding macromolecular
environment. See the section
Calculation of MMFF94 Binding Enthalpies
in the appendix for further details.
Minimize MMFF94 Energy
minimizes the MMFF94 energy of library molecules whether the library molecules
are located in a binding site (Settings scheme: "Binding Pocket Molecule Energy Minimization")
or are solely available (Settings scheme: "Free Molecule Energy Minimization").
To customize the settings, see the section
Force Field: MMFF94
and
Force Field: Minimization
in the preferences.
Calculate Interacting Feature Counts
calculates the number of interacting features between library molecules
and the binding pocket. The distribution of structure-based pharmacophoric
features of each library molecule is shown in the library view. This is useful
to validate e.g. docking poses.
Clear User Properties
removes all user
calculated properties of the library table.
Clear File Properties
prevents the
display of all library properties that are contained in
a loaded
file.
Add Molecules
adds additional molecules
to your Training-Set, Test-Set or set of Ignored Ligands.
Flag Selected Molecules
marks selected molecules as Training-Set, Test-Set or
set of Ignored Ligands. You can also decide, which molecules
are active or inactive.
Remove non-Espresso Properties
deletes external properties of the Ligand-Set, that are not generated by Espresso.
Remove Pharmacophore Bias
deletes the
user-defined pharmacophore that influences the ligand-based pharmacophore generation.
(Re)Create Conformational Models
creates conformations to your Ligand-Set. If conformations already
exist, they are recreated.
Cluster Ligand-Set
clusters
the Ligand-Set by pharmacophore based ligand similarity.
Create Pharmacophore
automatically creates the pharmacophore, that is shared in the
Training-Set and matches it with the Test-Set. Conformations are
automatically generated for the molecules if not available.
Re-Interpret Ligand
(CTRL+R)
causes LigandScout to re-interpret all bonds of the
ligand shown in the current
Active Site View
.
This also triggers recalculation of properties like
stereo chemistry, hydrogen positions, and aromaticity.
Move Selection to Ligand Side
(CTRL+SHIFT+C)
adds the selected environment elements to the ligand side.
LigandScout considers these elements as part of the ligand in
pharmacophore generation. It is also possible in the
Macromolecule View
to select some amino acids or nucleic acids and move
them to the core yielding new ligands instead of the acids bound to
the macromolecule.
Move Selection to Macromolecule Side
(CTRL+SHIFT+E)
adds the selected ligand elements to the macromolecule side.
LigandScout considers these elements as part of the environment in
pharmacophore generation. This entry will only be active in
Active Site View
.
Create New Bond
(CTRL+SHIFT+B) adds a new bond between
two selected atoms if it is
reasonable
.
Essentially this means that two atoms can be bound together if
their distance is not too short and not too long. This distance
should be around the two atoms "normal" bonding distance.
Increase Charge of the Selected Elements
(+)
adds a positive charge to the currently selected atoms.
Decrease Charge of the Selected Elements
(-)
adds a negative charge to the currently selected atoms.
Deprotonate all single-bonded oxygens of all acid-like
substructures (CTRL+SHIFT+J). For nitrogens which are double-bonded to two oxygens
one of the double-bonds will be replaced by a single bond, the
nitrogen's charge will be set to
+1
and the oxygen will be deprotonated. Basic amines, amidines, and
guanidines are protonated.
For all single-bonded deprotonated oxygens of acid-like
substructures and all nitrogens which have a double bond to an
oxygen and a single bond to a deprotonated oxygen, the deprotonated
oxygens will be set back to a neutral state (protonated). Basic
amines, amidines, and guanidines are neutralized (CTRL+SHIFT+K).
Move All Water Molecules and Metals to
Ligand Side
(CTRL+SHIFT+L) relocates all metals and
water molecules to the ligand side.
Move All Water Molecules and Metals to
Macromolecule Side
(CTRL+SHIFT+M) relocates all metals and water
molecules to the macromolecule side.
Minimize MMFF94 Energy of Molecule
(CTRL+ALT+O)
minimizes the MMFF94 energy of the ligand. For an unbound ligand the
MMFF94 and optimization settings scheme “
Free Molecule Energy
Minimization
” applies. For a ligand within a binding pocket the
settings scheme “
Binding Pocket Core Molecule Energy Minimization
”
is used. To customize the settings, see the section
Force Field: MMFF94
and
Force Field: Minimization
in the preferences.
Minimize MMFF94 Energy of Side Chains
(CTRL+ALT+SHIFT+O)
minimizes the MMFF94 energy of the side chains of the environment.
The settings scheme “
Binding Pocket Side Chain Energy Minimization
” is used.
To customize the settings, see the section
Force Field: MMFF94
and
Force Field: Minimization
in the preferences.
Minimize MMFF94 Energy of Core Molecule and Side Chains
(CTRL+ALT+SHIFT+X)
minimizes the MMFF94 energy of the ligand and the side chains of the environment.
The settings scheme “
Binding Pocket Core Molecule and Side Chain Energy Minimization
” is used.
To customize the settings, see the section
Force Field: MMFF94
and
Force Field: Minimization
in the preferences.
Generate Tautomers
(CTRL+ALT+G)
performs a tautomer enumeration of the shown ligand.
Create new Feature
can create
three geometric types of features: vector (H-Bond Acceptor, H-Bond Donor,
Iron Binding Location, Magnesium Binding Location, Zinc Binding Location),
point (Hydrophobic, Negative Ionizable, Positive Ionizable, Exclusion
Volume) and plane feature (Aromatic Ring). To create a vector feature between
the ligand and the environment, select an atom on ligand-side and on
environment-side. Then, you can choose your appropriate feature.
To create a feature only on the ligand-side, select at least one
atom of the ligand. To create an aromatic ring feature, at least three
atoms are needed to be selected. If you choose more than one atom
(ligand-side only), the new feature is created on the average
position of the selected atoms.
Change Feature Type
alters the type
of a feature. Select a feature and change its type. A submenu
shows which types can be chosen.
Create Pharmacophore
(CTRL+F9)
activates LigandScout's automatic pharmacophore generation
algorithm. Both ligand and environment characteristics are analyzed
for pharmacophore generation.
Create Simplified Pharmacophore (Catalyst)
(SHIFT+F9) reduces
advanced 3D pharmacophore models
for
virtual screening with Catalyst
. In case of multiple
features on single atoms, LigandScout automatically obtains the best feature for Catalyst.
Create Simplified Pharmacophore (MOE)
(CTRL+SHIFT+F9)
reduces
advanced 3D pharmacophore models
for
virtual screening with MOE
. LigandScout will prepare
the pharmacophore in a way which works best for MOE.
Create Simplified Pharmacophore (Phase)
(CTRL+SHIFT+F10)
reduces
advanced 3D pharmacophore models
for
virtual screening with
Phase
. LigandScout will prepare the pharmacophore in a way
which works best for Phase.
Create Exclusion Volumes Coat
places
individually adjusted exclusion
spheres
on the ligand surroundings occupied by protein residues. This
feature allows one to simulate the binding site shape in order to
significantly increase enrichment during virtual screening.
Create Exclusion Volumes on Selected Atoms
places exclusion volumes on selected atoms.
Increase Selected Feature Tolerance by 0.15
Å
(CTRL+SHIFT+NumPad+) expands the
tolerance spheres
of pharmacophoric features by 0.15 Å in
order to decrease the restrictivity of the pharmacophore model.
Decrease Selected Feature Tolerance by 0.15
Å
(CTRL+SHIFT+NumPad-) narrows the
tolerance spheres
of pharmacophoric features by 0.15 Å in order to increase
the restrictivity of the pharmacophore model.
Increase Selected Feature Weight by 0.1
rises the weight of a selected feature.
The feature weight has an impact on the pair-wise alignment of pharmacophores.
(maximum: 1)
Decrease Selected Feature Weight by 0.1
lowers the weight of a selected feature.
The feature weight has an impact on the pair-wise alignment of pharmacophores.
(minimum: 0.1)
Interpolates selected features in the
Alignment Perspective
(CTRL+E).
The features
must be of the same
type
and
must belong to the same
pharmacophore
to yield a new chemical feature replacing
the selected old ones. Features will only be interpolated if their
maximum pairwise distance will be lower than five times the average
tolerance of the selected features.
Mark Features as Optional
flags the
selected features which are not mandatory to find a valid alignment
(similar to the max. omitted feature action). During the screening process,
a molecule is still a valid hit when these marked features are not matched.
Unmark Features as Optional
reverses the flag of
the selected features. To switch off features, select
Mark Features as Disabled
. These features are not
taken into account in the alignment process. To switch disabled features on,
select
Mark Features as Enabled
.
Use this functionality if you want to screen the
current pharmacophore model against a molecule library which
is not loaded in LigandScout. The
Open
dialog
appears where you can choose the library and customize the
settings
for the screening procedure
.
Centers all shown elements in
Alignment Perspective
(CTRL+ALT+C).
Sets the selected ligand or pharmacophore explicitly as
reference for the alignment procedure in the
Alignment Perspective
(CTRL+ALT+R).
Clear All Elements
(CTRL+ALT+X) deletes all molecules
and pharmacophores which are currently listed in the
Alignment Perspective
.
An confirmation pop up reconfirm your decision.
Aligns the selected elements to the reference element
(
Align by features
) or with respect to the protein environment
(
Align by reference points
) in the
Alignment Perspective
.
Align by features
allows one to align both ligands and pharmacophore
models by an algorithm examining the best possible pharmacophoric
feature overlay. If no reference element was specified one of the
selected elements will be chosen randomly.
Align by reference points
is a useful tool to investigate
protein-ligand complexes of structurally related targets. It allows one
to align the ligands with respect to the surrounding protein
residues. In this way the user is able to directly compare the
absolute position of one or several ligands within the aligned
binding site.
Allows one to generate
shared
feature pharmacophores
based on
feature alignment
or
reference points alignment
.
Generates
merged pharmacophores
based on
feature alignment
or the
alignment of reference points
of the selected pharmacophores and the pharmacophores of the selected molecules in the
Alignment Perspective
. Every single chemical feature will be added (with its
original positions) to a newly created pharmacophore, the so-called
merged pharmacophore
.
Joins
all features
of all selected pharmacophore models in one
pharmacophore (CTRL+ALT+M).
In the
Alignment Perspective
this action will automatically group features of the
same type which are within their mutual tolerances
(CTRL+ALT+T). Every group of
features will be independently interpolated yielding new features.
These new features will replace the original groups of features in
the resulting pharmacophore. In other words, the resulting
pharmacophore will not contain features with an overlap of
their tolerances that is too large.
The
Ligand Default Style
menu provides
access to a variety of rendering modes, like
Line
,
Ball and Stick
,
Stick
and
Spacefilling/CPK
. Hydrogens can be excluded
from depiction or be reduced to stubs.
Show
Templates
activates the depiction of default geometric
templates depending on the hybdridization-state of the currently
selected atom.
Show Interactions
displays all
possible interaction partners within the macromolecule of the
currently selected ligand atom.
Show Ionic
Interactions
displays ionic interactions between metals
and non-metals. The
Pickable
check box sets the
ligand selectable/unselectable.
The
Environment Default Style
submenu of the
Render Control
menu corresponds
in large part to the respective ligand menu and
lets you select different rendering modes and define hydrogen
handling.
Show Templates
activates the
depiction of default geometric templates depending on the
hybdridization-state of the currently selected atom. The
Pickable
check box sets the environment
selectable/unselectable in
Active Site View
.
Selection
lets you customize the
rendering of the current selection. LigandScout provides four
different modes:
Line
,
Ball and
Stick
,
Stick
and
Spacefilling/CPK
.
The
Protein Backbone Default Style
submenu lets you control the rendering of the protein backbone.
LigandScout provides four different modes:
Lines
,
Splines
,
Snake
, and
Ribbon
.
The
Pharmacophore
submenu enables you to
switch on/off pharmacophore and excluded volume rendering. The
Pickable
check box makes the pharmacophoric features
selectable or unselectable in 3D.
Use Blur (SDOF: Semantic Depth of Field)
allows advanced blurring of objects. The effect is only visible if
the
corresponding
check box
of ligands/pharmacophores in the Alignment Panel
is activated. SDOF may cause graphical issues or slow down the
performance on low end computer systems and is therefore
recommended only with suitable hardware equipment.
Show Reference Points
allows to visualize
the position of the protein residues when using
alignments based
on reference points
.
Show Reference Point
Names
places residue identifiers to this reference
points.
Show Feature Tethers
visualizes the
affiliation of corresponding pharmacophoric features with tethers.
Show Reference Point Tethers
visualizes the
affiliation of corresponding protein reference points with
tethers.
The
Behavior
submenu gives you control
on the main
3D View
defaults.
Pickable Bonds
toggles the possibility to select bonds.
Highlight Bond
Cylinders in Selection
will only show an effect in the
render styles
Ball and Stick
and
Line
. If this option is disabled only the atom
will be highlighted in
Ball and Stick
render
style and nothing will be highlighted in
Line
render style. However, if this option is enabled a selected atom
will also highlight all “
outgoing
” bonds half-way for both render
styles.
Simple Rendering While Moving
(CTRL+V) causes
LigandScout to use less complex rendering algorithms while moving
the macromolecule-ligand complex.
Show
Tooltips
shows you important information, such as
classification, geometry, and coordinates of your currently
selected atom, bond, or pharmacophore feature. Enabled option
causes LigandScout to view a small information popup next to the
selected object.
LigandScout supports parallel and perspective projection.
Wide Angle View
enhances perspective
distortion. The
Light Background
option (CTRL+L)
switches the 3D depiction to white background.
Depth
Cueing
enhances depth recognition in the scene.
AntiAliasing
neutralizes stairway-effects at
the edge of rendered objects.
Center Camera on Ligand
(CTRL+Page Up) recenters the
camera position on the ligand.
Center Camera on Binding Site
(CTRL+Page Down) recenters
the camera position on the protein binding site.
Turn Camera for Optimized View
recenters
the camera position. This feature allows to quickly reposition the
protein-ligand complex with minimum overlap of the ligand
atoms.
Allows one to measure the angle between three selected atoms. You
can erase the angle monitor by selecting the angle value and
pressing the
Delete
icon.
Allows one to measure the distance between two selected atoms.
You can erase the distance monitor by selecting the distance value
and pressing the
Delete
icon.
Depending on the van der Waals distances of ligand atoms
spheres will be created and combined to the van der Waals
surface (CTRL+ALT+SHIFT+W).
This surface describes the boundary which separates the
ligand from its potential interacting atoms (CTRL+ALT+SHIFT+A). This means, points on
this surface are the closest locations, where interacting atoms can
appear.
This surface is created by placing a sphere around each
ligand atom (CTRL+ALT+SHIFT+M). Then a hypothetical probe is rolled over these
spheres. The record of where this probe touched the spheres will
result in the molecular surface.
This will display the surface of all the atoms in the
environment which potentially interact with ligand atoms
(CTRL+ALT+SHIFT+B). However,
only those parts of the surface facing the ligand will be
drawn.
This option will toggle the visibility of parts of the
surface which will be seen from behind. This is especially useful
for viewing a receptor surface, because this will allow you to cut
away the front part of the enclosing view and show the ligand as
illlustrated below:
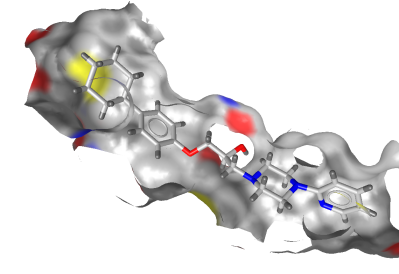
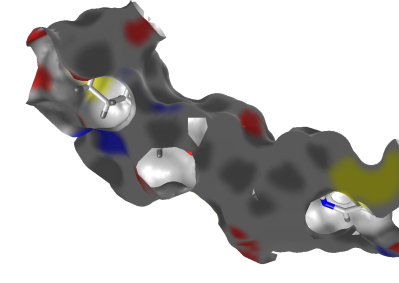
Activating this option will result in a surface rendered
transparently such that for example also the interior of an
enclosing surface will be visible.
This option allows the fully automated recalculation of
molecular surfaces upon molecule changes. The prerequisite for this
option to have an effect is that a molecular surface type other
than “
None
” is selected.
Front clipping
suppresses the
visualization of parts of the surface that may avert a direct view
of the ligand in the binding site. This feature is especially
useful in the case of tight, elongated pockets.
This menu item allows for coloring the surface. Available
options are:
No Color-Coding (Gray)
,
By Atom Type
,
By
Polarity
,
By Aggregated
Lipophilicity/Hydrophobicity
and
By MMFF94
Partial Charge
.
-
No Color-Coding (Gray) no specific coloring (defaults to gray).
-
By Atom Type will use different colors for each atom type.
-
By Polarity will color polar regions in red (hydrophilic areas).
-
By Aggregated Lipophilicity/Hydrophobicity will color non-polar regions in yellow (hydrophobic areas).
-
By MMFF94 Partial Charge will color by MMFF94 partial charge properties.
This menu item allows to choose between different render
styles like solid, wireframe or rendering points.
Depending on the perspective you
can enable/disable the
PDB Control
,
Library/List
,
Alignment
and
Bookmark Panels
. The
PDB Control Panel
gives you information about the current PDB file and lets you
select a ligand in the
Active Site View
.
The
Library/List Panel
shows
molecules contained in a library. The
Alignment Panel
allows you to align ligands and pharmacophores. In
the
Bookmarks Panel
you can save the current
Active Site View
.
The
2D View Panel
shows the 2D structure
and pharmacophore of the currently selected ligand
(
Ligand 2D View
). Important information and
statistics are shown as
Ligand Details View
in
Structure-Based Modeling Perspective
and
Alignment Details
View
in
Alignment Perspective
.
You can switch the
2D View Panel
on/off
by using the keyboard shortcut CTRL+2.
The
Hierarchy View
(CTRL+T)
gives a clearly arranged tree list of the currently
visible elements (either the whole macromolecule or the active
site, ligands and pharmacophores). You can also toggle visibility
and colorize elements of the
3D View
.
The
Library View
allows to examine an external compound library of
small organic molecules. It is especially useful for the
investigation of docking results in the
Structure-Based Modeling Perspective
.
The
Ligand-Set Table
shows the ligands (Test-Set, Training-Set or Ignored Ligands) that are used
for the ligand-based pharmacophore generation in the
Ligand-Based Modeling Perspective
.
The
Screening Hits
shows the hits of a screening run in the
Screening Perspective
.
Enables/Disables several types of widgets
to exchange and navigate through your data. The
Copyboard Widget
transfers ligands, pharmacophores and active sites between perspectives. The
Zoom Out Widget
switches from the
Active Site View
to the
Macromolecule View
. The
Alternatives Switcher
helps you to go through your collection of selected compounds in the Library View.
The
Tautomer Switcher
and the
Conformer Switcher
guides you through the
tautomers or conformers of a specific compound.
Show All Views and Tables
(CTRL+SHIFT+A) enables all
panels and control windows depending on the current view
(
Ligand-Based Modeling Perspective, Structure-Based Modeling Perspective, Alignment Perspective, Bookmark View
or
Screening Perspective
).
LigandScout Help
(F1) starts the built-in
help system. The chapters describes the functions that are provided
by LigandScout. You can also search specific terms in the
Help
dialog.
Displays information
about LigandScout and the development team. The corresponding entry is
located in the
LigandScout
menu on Mac OSX
platforms.